Export ortholog groups as protein or nucleotide sequences¶
Note
Data availability for this tutorial: The data required to complete this tutorial include:
This tutorial demonstrates how to export ortholog groups from a previous Orthology search operation as protein and nucleotide sequences.
Load group files¶
Let’s import the results from the previous search of orthologs across 10
genomes (see tutorial
Basic search of orthologs among 10 proteome files).
Navigate to the Orthology screen, Explore section, and click the
+ button at the top left of the screen. Go to the directory containing the
group files from the corresponding ortholog search operation and select
one or more files. Here we’ll select only one. Once loaded, the basic
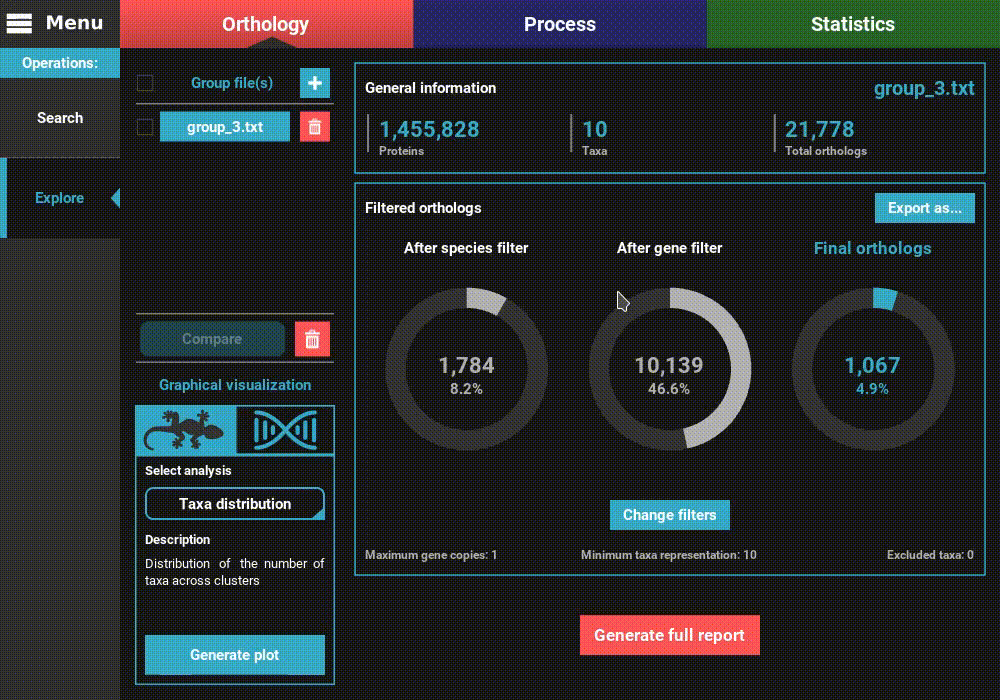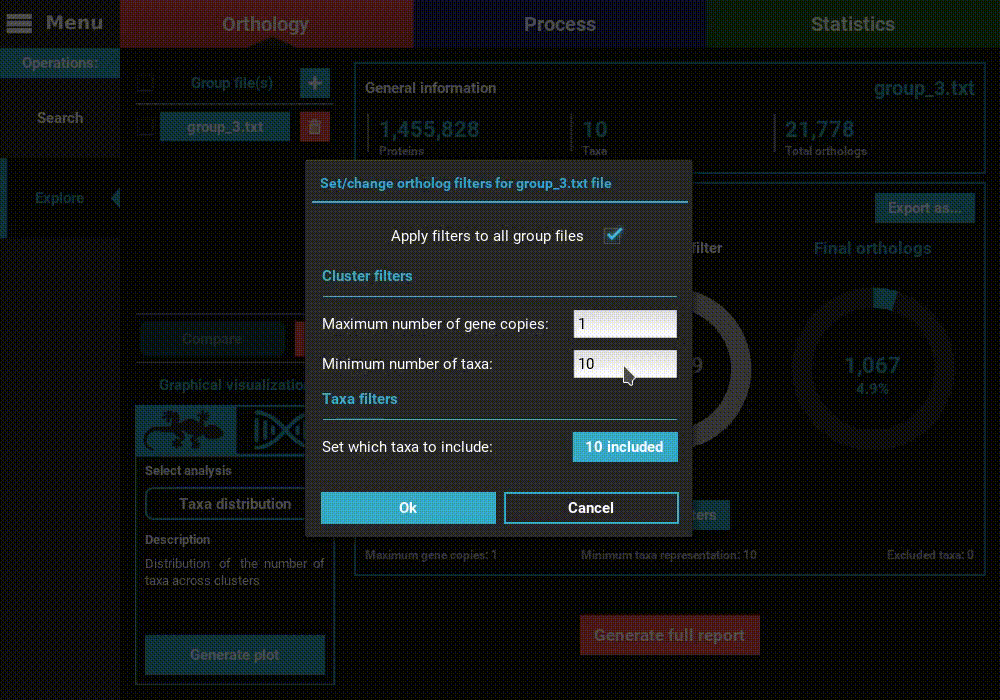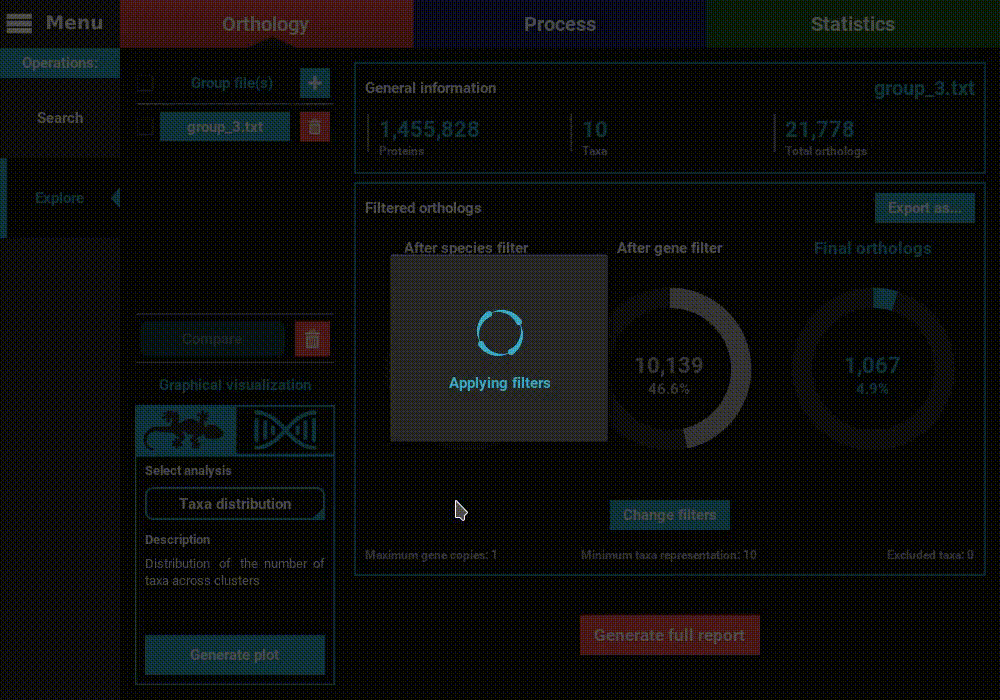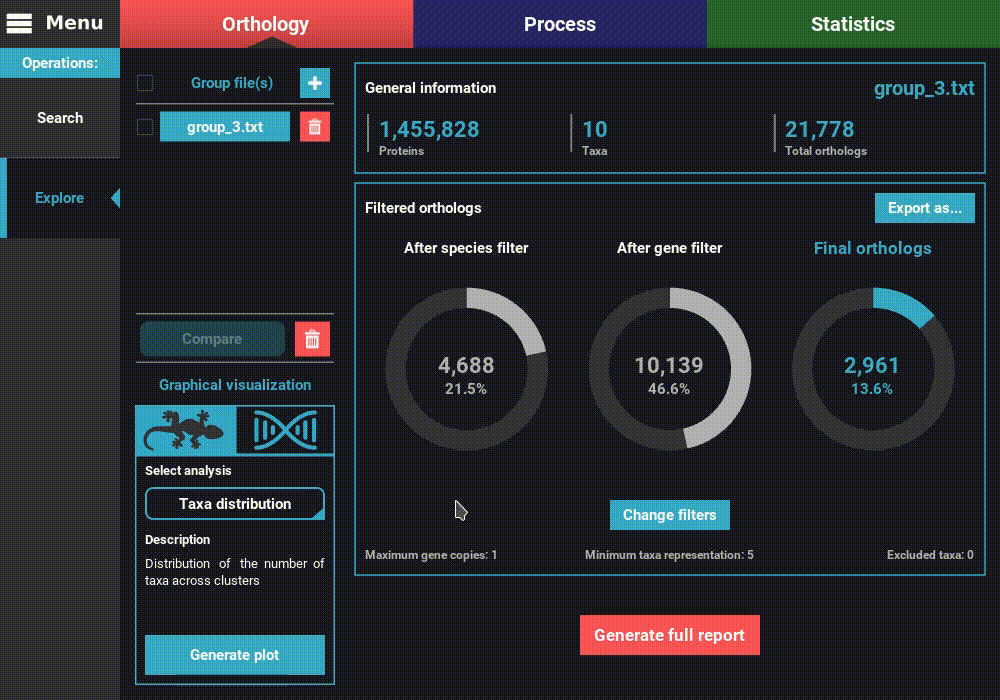
information of the group file will be displayed for the default
orthology filters (only single copy genes present in all species).
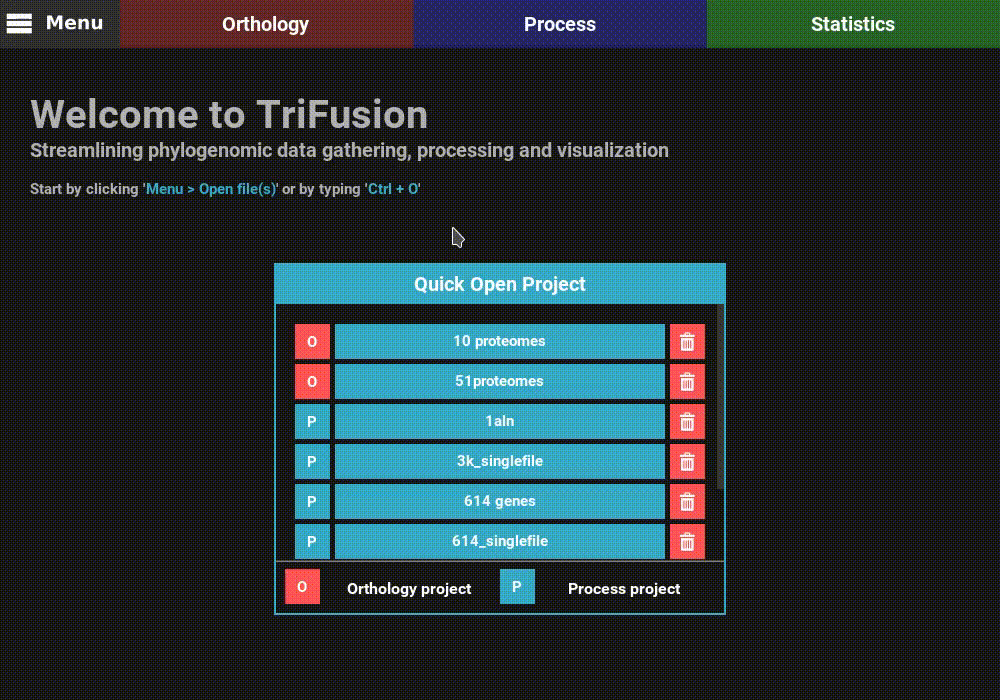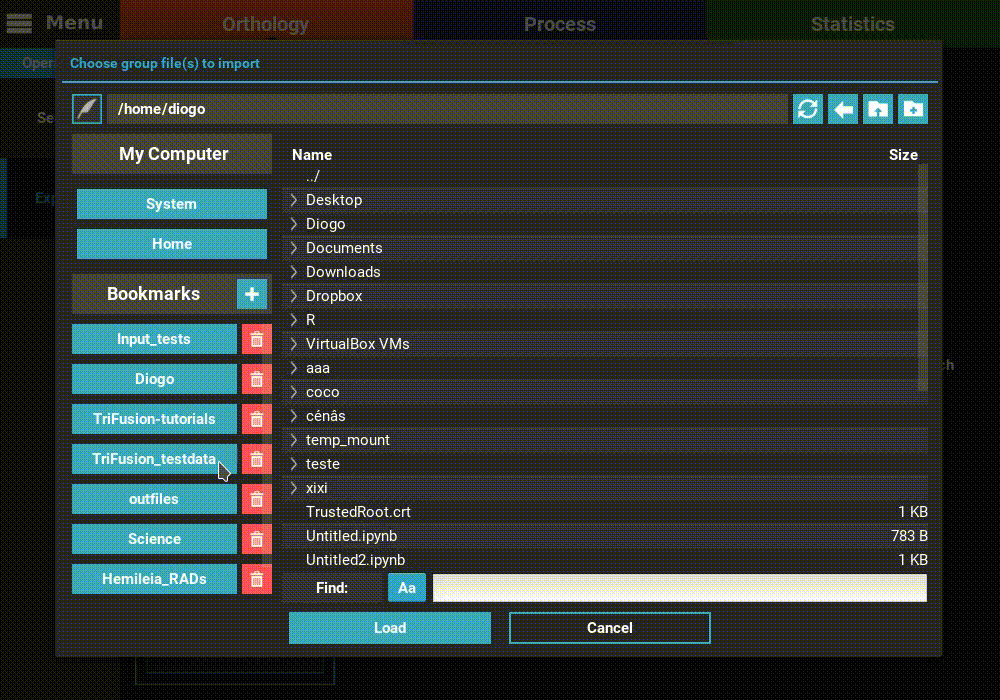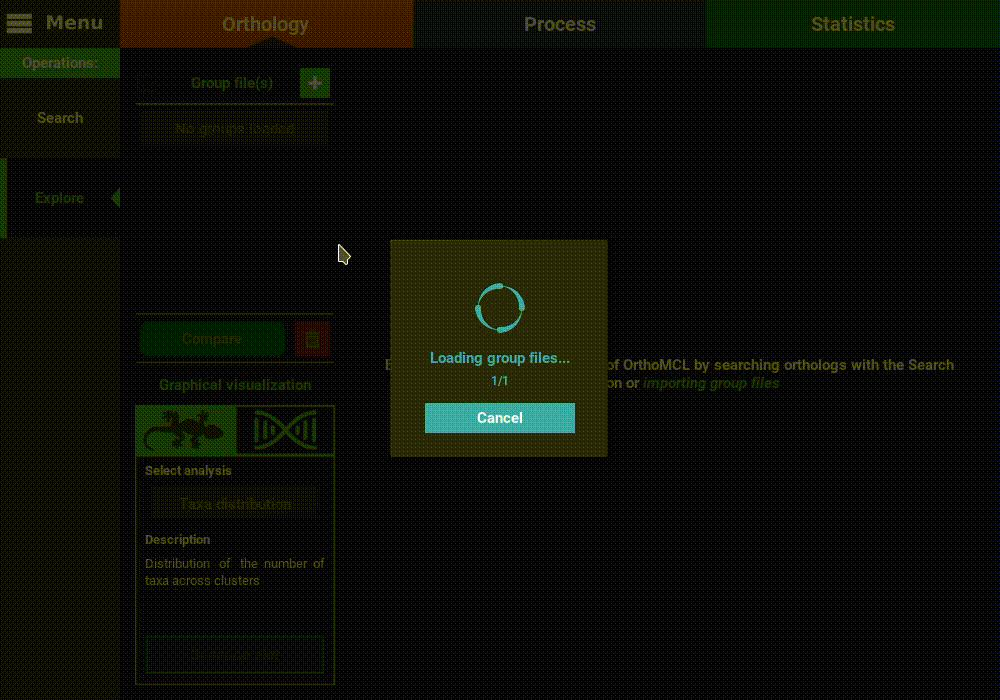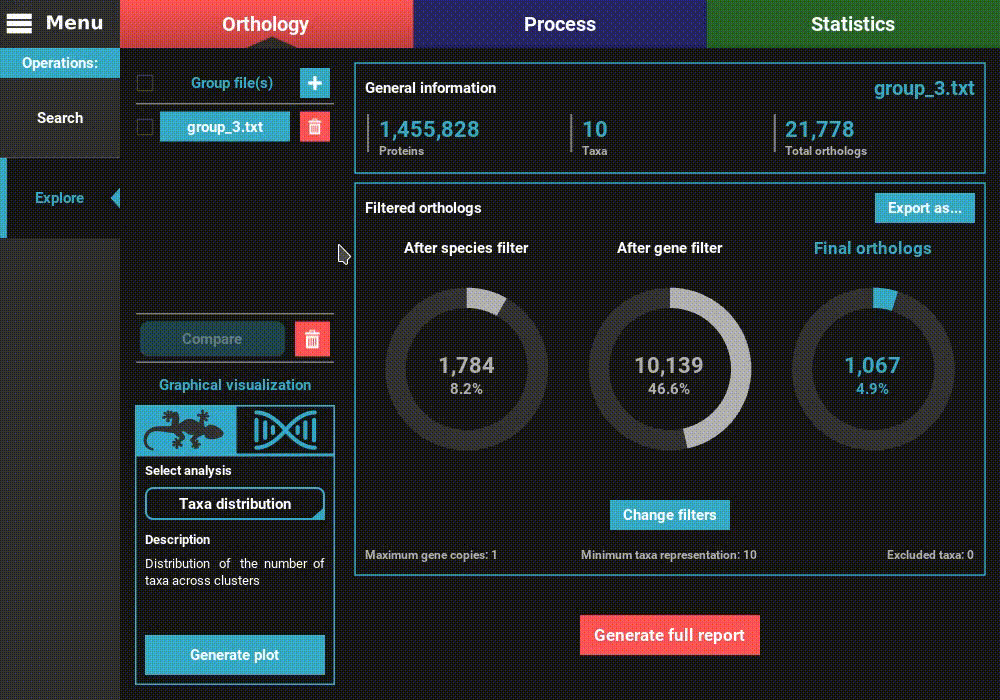
However, let’s change the filters for something more permissive in terms
of minimum taxa representation. Click the Change filters button, and
change the minimum number of taxa value to 5 (50% of taxa representation).
Click Ok and the information on the screen should be updated to
something like this.
Export into protein sequences¶
First click the Export as... button in the Explore section screen.
This will open the export group dialog. To export the ortholog groups into
protein sequence files (in Fasta format), a protein database of all
input genomes must be provided. This file is automatically generated during
the Orthology search operation and is stored in the backstage_files
directory, with the default name of goodProteins_db (this name can
be change by the Database name option). If you have just finished an
Orthology search operation in the current session of TriFusion, this
database file is already set. However, if you are executing a different
session of TriFusion, you’ll need to provide this file.
A protein database file is simply a Fasta file that contains all sequences used during the ortholog search procedure, with simplified headers. TriFusion will look for the sequence headers in the groups file and fetch the corresponding sequence from this database file.
Click the Protein sequences button. This will make the Protein database
base option available. To search and select the database file,
click the Select... button.
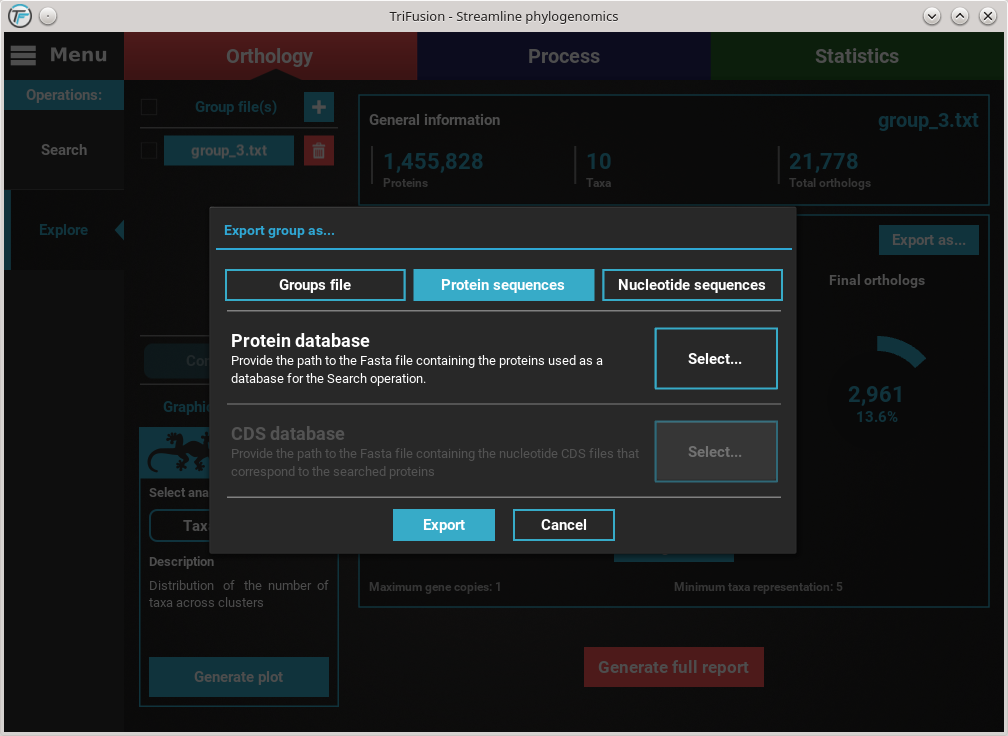
Notice that I navigated to the results directory of my previous ortholog
search and then to the backstage_files directory. Since I did not change
the Database name option value in TriFusion, I have a goodProteins_db
file in this directory. If you are using the downloaded tutorials data,
select the protein database file. Then click Save.
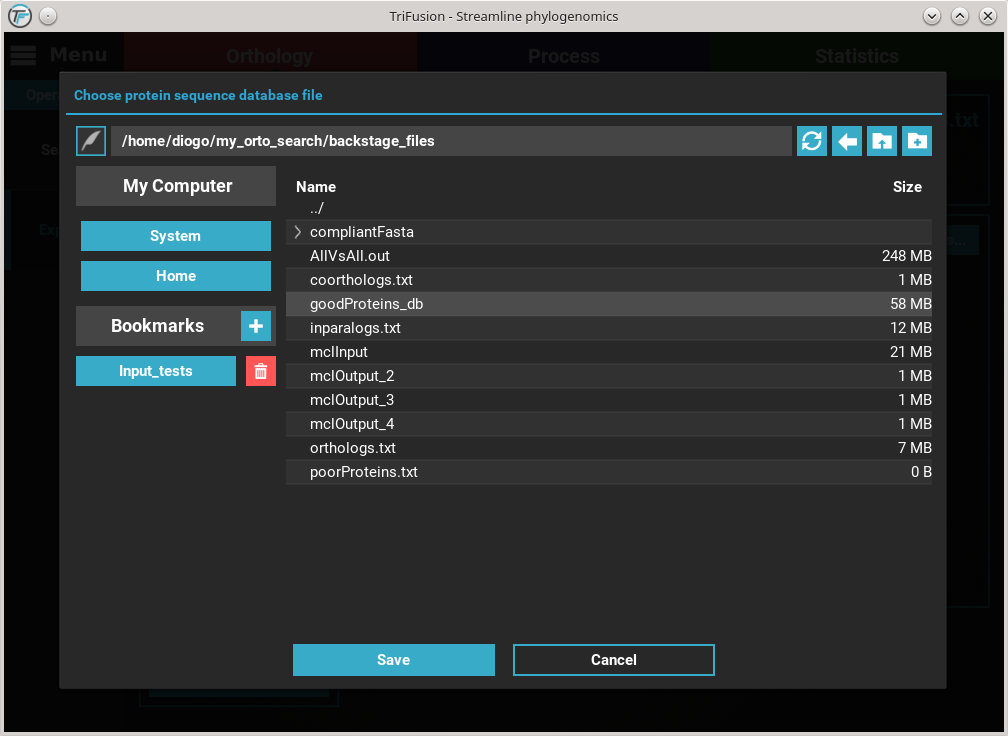
You’ll notice that the Protein database button changed in accordance to the
name of the protein database file. Finally, to export the ortholog groups
click the Export button. Select or create a directory where the new
files will be generated and then click Ok. At the end of the export
operation, a success popup should appear informing the number of
ortholog groups exported.
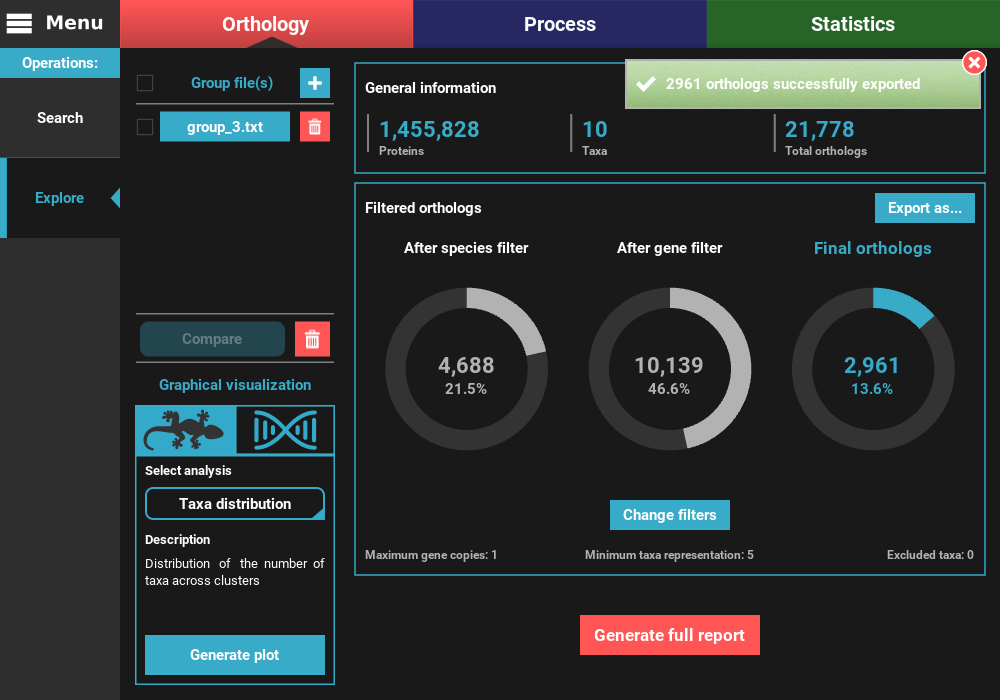
Your protein sequence files are ready to be used in the specified directory. Notice that TriFusion will set the same name for each taxon/species across the protein sequence files. For instance, sequence references from a given species in multiple ortholog groups of Necoc|153 and Necoc|646 will be appear as Necoc in all sequence files. The correspondence between each taxon sequence and the original header in the groups file will be written in the header_correspondance directory, for each protein sequence file.
Export into nucleotide sequences¶
Note
To export ortholog groups, a working executable of USEARCH is required. See the Setup of USEARCH tutorial.
First click the Export as... button in the Explore screen. This
will open the export group dialog. To export the ortholog groups into
nucleotide sequence files (in Fasta format), a protein database AND
cds/transcript files must be provided.
The CDS/transcript files are usually associated with the proteome files in genome sequencing projects.
Click the Nucleotide sequences button. This will make available the
Protein database and CDS database options.
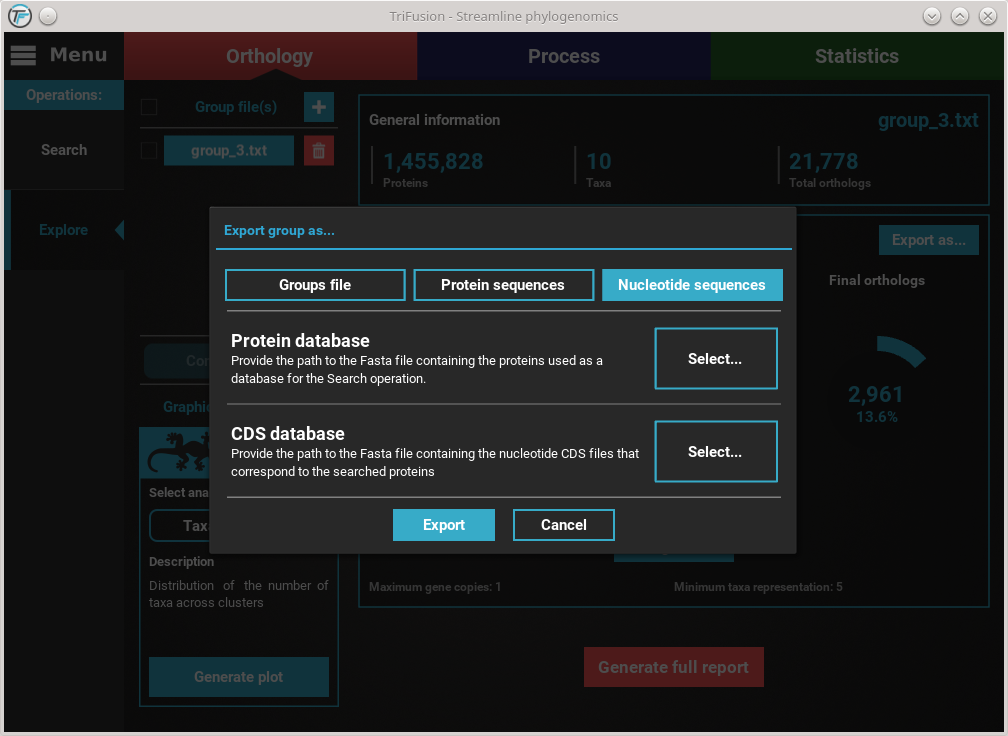
Refer to the previous Export into protein sequences section on how to
set the protein database file. After setting this file, the
cds/transcripts that correspond to the proteomes used during the
Orthology search operation, must be also provided. You can have
an individual cds/transcript file for each species, or concatenate all
files into a single master file. Click the Select... button of the
CDS database option and search for the cds/transcript files. If you
are using the tutorial’s material, provide the
CDS files.
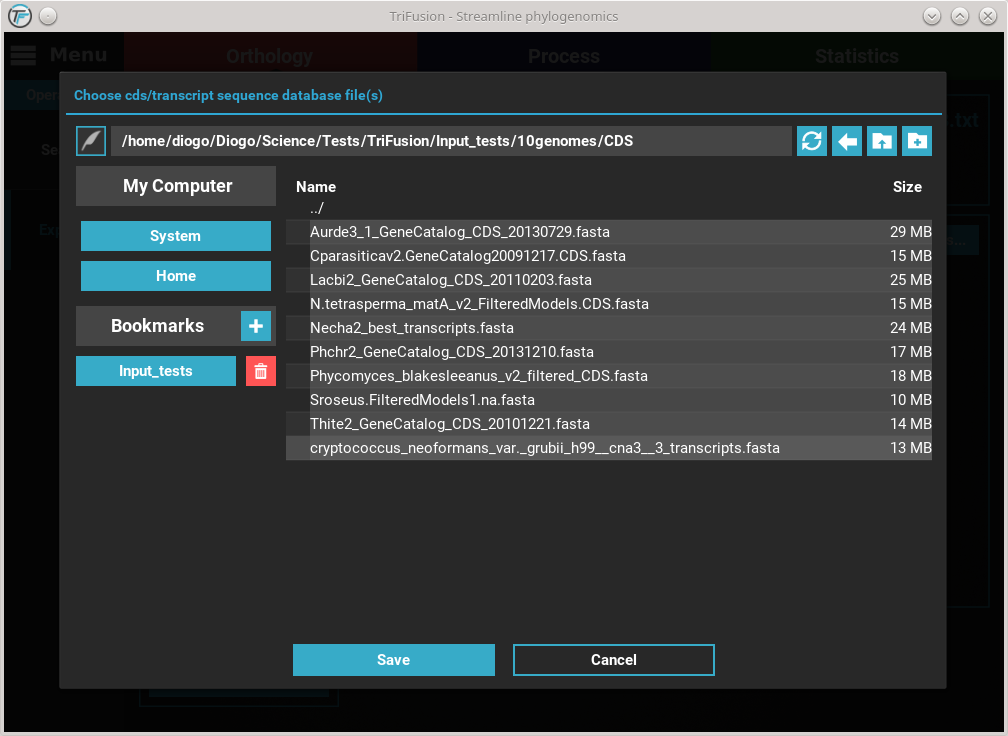
Here, I have the CDS and transcript data for each of the 10 species in
their respective individual files. Select them all with shift + click
and click Save. You should notice that the CDS database button changed
in accordance to the number of files select, which is 10 in this case.
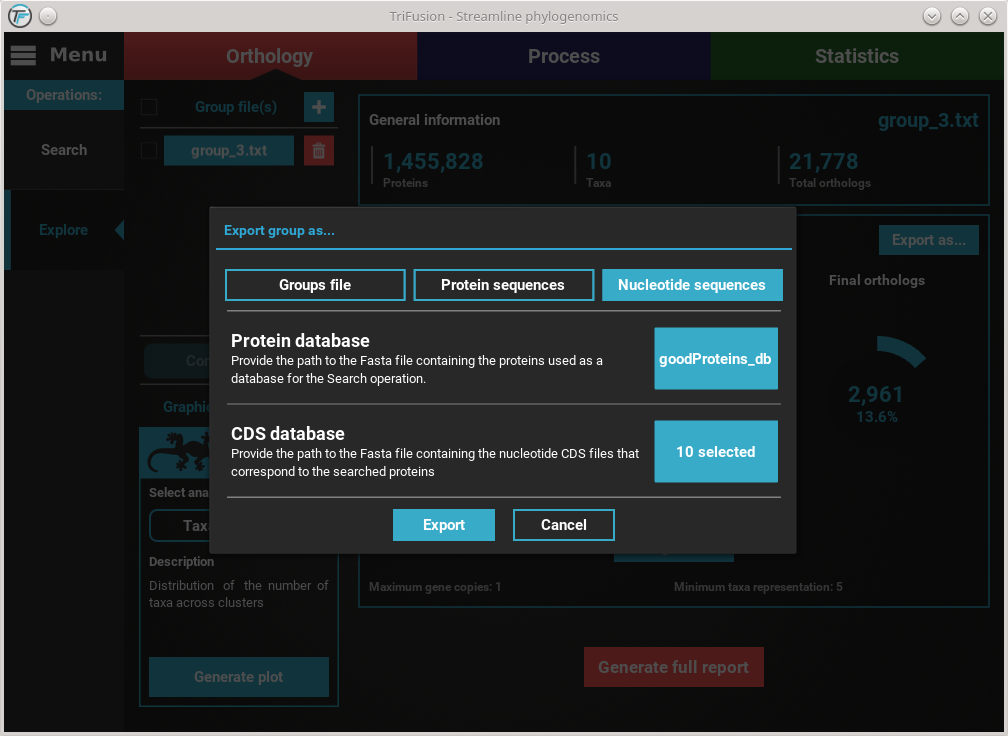
With both the protein database and cds/transcript files selected,
we are ready to begin the ortholog export. Click the Export button and
select or create the directory where you want to generate the nucleotide
sequence files.
At the end of the export operation, a success popup should appear informing the number of sequences that were successfully exported.
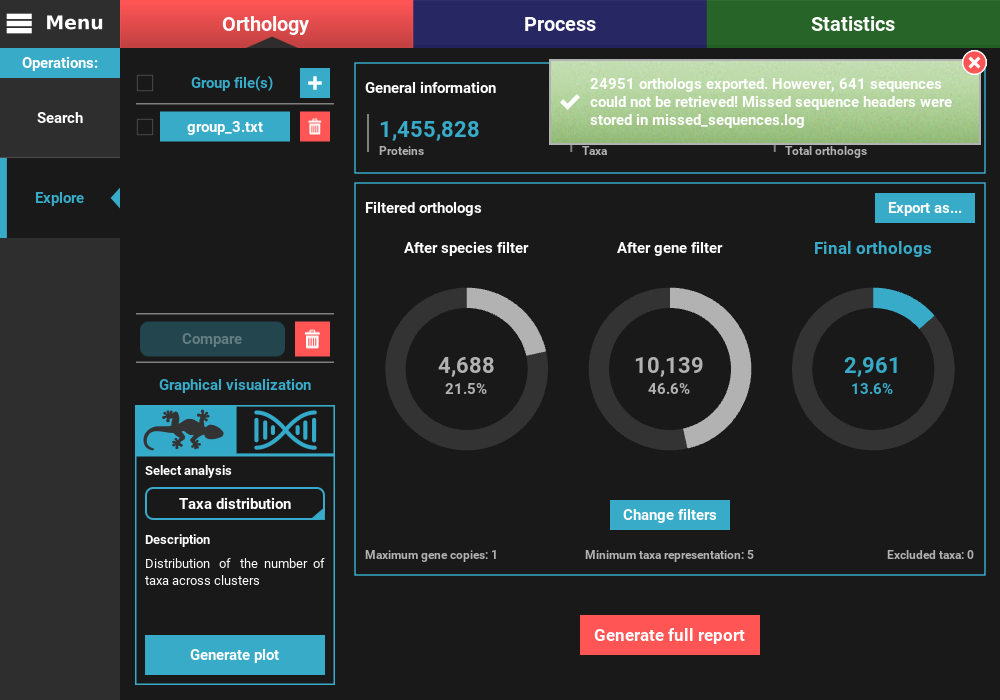
Note
Note on the sequences that could not be retrieved:
TriFusion converts groups into nucleotide sequences by searching the proteins from the main output of the Search operation in CDS/transcript databases provided by the user. The reason why this search is done instead of simply looking for sequence headers that are the same in the protein and nucleotide databases is because sometimes there is no such cross reference. Therefore, TriFusion creates two different databases and then uses USEARCH to search for perfect hits between the protein and nucleotide sequences. This ensures that the nucleotide sequences correspond exactly to the proteins referenced during the Orthology search operation. However, even with this method, some nucleotide sequences may be absent from the databases. Fortunately, this represents only a minority of the cases. In this example, 641 protein sequence had no match in the nucleotide databases provided by the user, which represents only 2.8% of the total dataset. In most cases, this occurs only on a limited number of species but in any case, make sure that the proteome and CDS/transcript files correspond to the same version of the genome sequencing project.