Load data into TriFusion¶
TriFusion deals with different types and formats of input files, depending on which module you want to use. The Orthology module deals with proteomes and group files while the Process and Statistics module deals with alignment files. Regardless, input files are loaded into the application mostly in the same way (see How to load data into the app below).
Input types and formats¶
Orthology - search¶
Proteome files can be provided as the input for the Orthology search
operation. These are Fasta formatted files, each with the amino acid
sequences of a single species. TriFusion will interpret the name of the
proteome file (minus extension) as the taxon name, so it is recommended that
these files are named accordingly (for instance, Aspergilus_fumigatus.fasta
will appear as Aspergilus_fumigatus in the final ortholog files).
The only requirements for the input files
is that the headers must have one or more fields separated by a “|” symbol,
and at least one of those fields must be different for all sequences.
A standard proteome file resulting from a genome sequencing project is usually something like this example of the fungus Aspergilus fumigatus:
>jgi|Aurde3_1|1209208|estExt_Genewise1.C_13_t10159
MTD(...)
>jgi|Aurde3_1|1326459|estExt_fgenesh1_pm.C_130053
MPP(...)
>jgi|Aurde3_1|1274305|fgenesh1_kg.13_#_18_#_isotig02263
MLY(...)
This is a valid input file for TriFusion with the headers containing 4 fields, the third one being the unique ID field.
Orthology - explore¶
Group files are one of the outputs of the Orthology search operation and the input of the Orthology explore operation. These are simple text files that contain all ortholog groups identified in the search operation by OrthoMCL:
Ortholog1: Afumigatus_proteins|433 Anidulans_proteins|4605 (...)
Ortholog2: Afumigatus_proteins|3278 Afumigatus_proteins|9183 (...)
Ortholog3: Anidulans_proteins|36 Anidulans_proteins|9893 (...)
(...)
Each line contains the name of the ortholog group and a list of sequence references separated by whitespace. Each reference (e.g., Afumigatus_proteins|433) corresponds to an actual protein sequence from one of the input protome files.
Process and Statistics¶
The Process and Statistics modules share the same input, which are sequence alignment files. The supported input formats are:
- Fasta
- Phylip
- Nexus
- Loci (PyRAD)
- Stockholm
The input format, sequence type (nucleotide or protein) and string formatting (leave or interleave) of the provided alignment files are automatically detected by TriFusion. The missing data symbol used in the input alignments will also be automatically detected from the three possible symbols of x, n or ?.
Note
Is there any constraint on how formats and sequence types can be loaded?
No. You can load files of multiple formats and sequence types all at once. All information will be automatically detected for each input alignments separately.
How to load data into the app¶
Note
Data availability for this tutorial: the small data set of 7 alignment files is available here.
Filechooser¶
Proteome and sequence alignment files can be loaded through the
application’s file browser. To do so, navigate to Menu -> Open/View Data
and click the Open file(s) button.
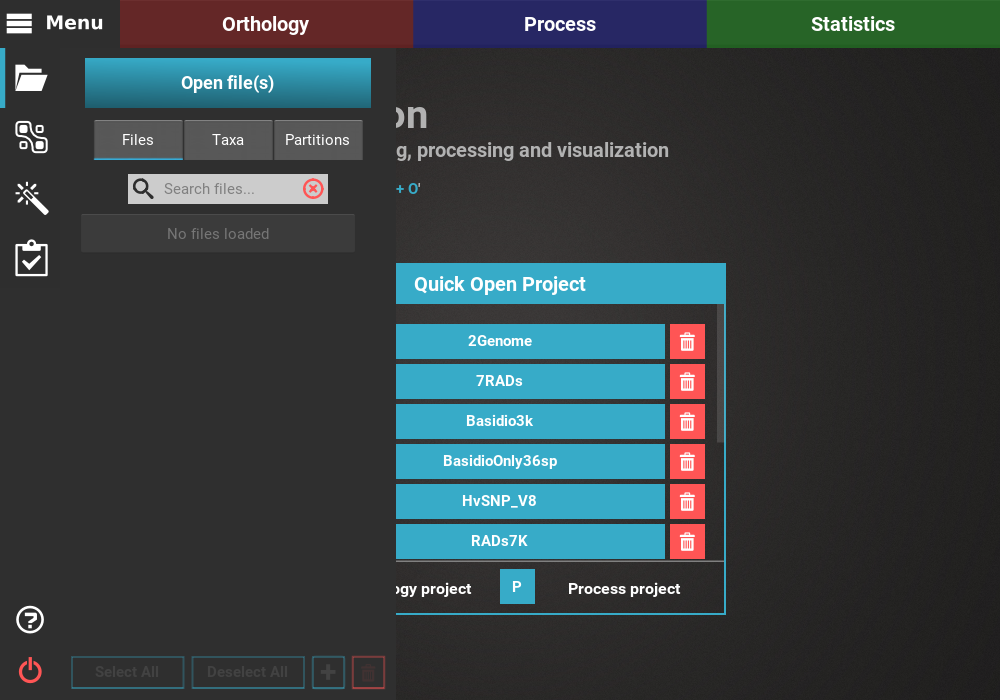
This will open the main file browser, which supports a couple of features:
- A list of bookmarks is displayed on the left, and any directory can be added to this list by opening it and clicking the
+button or pressingCtrl + D.- On the top of the screen, you can choose the input data type (whether you are loading proteome or alignment files).
- Below you can find the path of the current directory and several utility buttons to navigate the file browser.
- At the bottom of the file browser, there is a text field that searches folders and files in the current directory. There is also a drop down menu that filters files according to their extension.
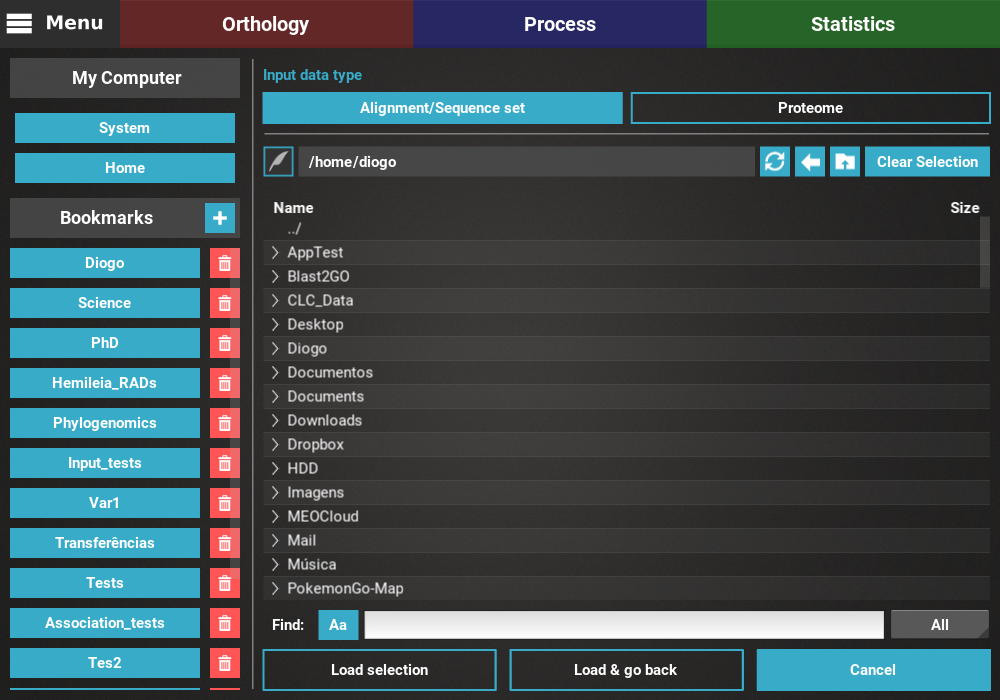
Navigate through the file browser by double clicking directories or clicking
on the > symbol. Multiple files can be selected by pressing either the
Ctrl or Shift keys. After completing you selection, click the
Load & go back button to load the data and go back to the previous screen.
If you wish to load additional data, click the Load selection button,
which will load the data but remain in the file browser screen.
In the example below, 7 files have been selected and are ready to be loaded.
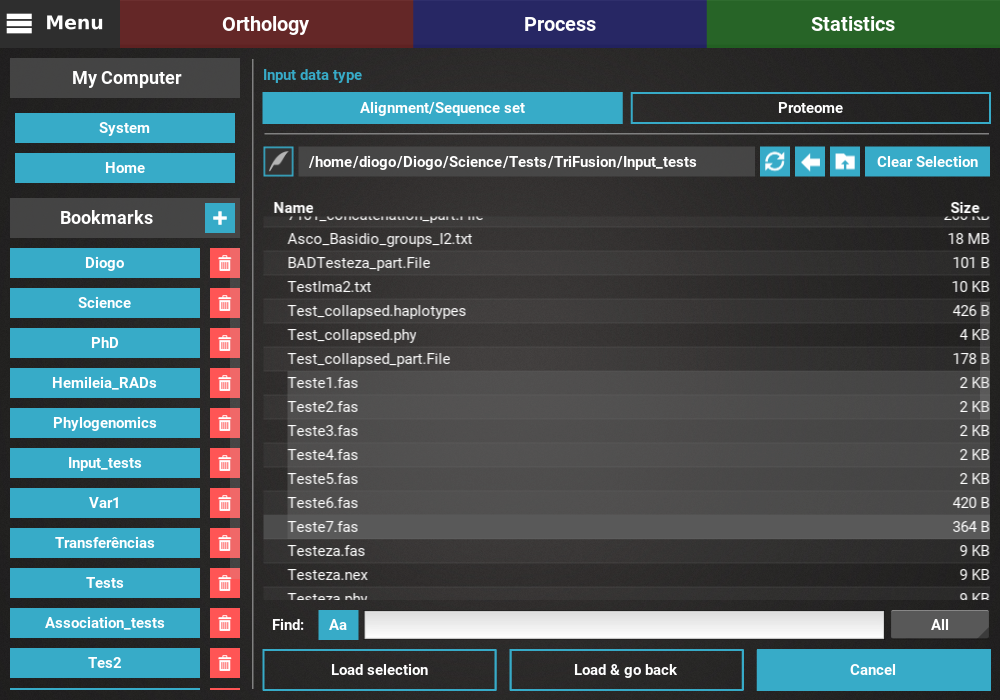
Note
TriFusion also supports the selection of one or more directories instead of files!
When directories are selected, all files contained in those directories will be loaded into TriFusion. If you are worried that not all files in a directory are alignments/proteomes, do not worry. TriFusion will ignore invalid input files while successfully loading valid alignment/proteome files.
Drag and Drop¶
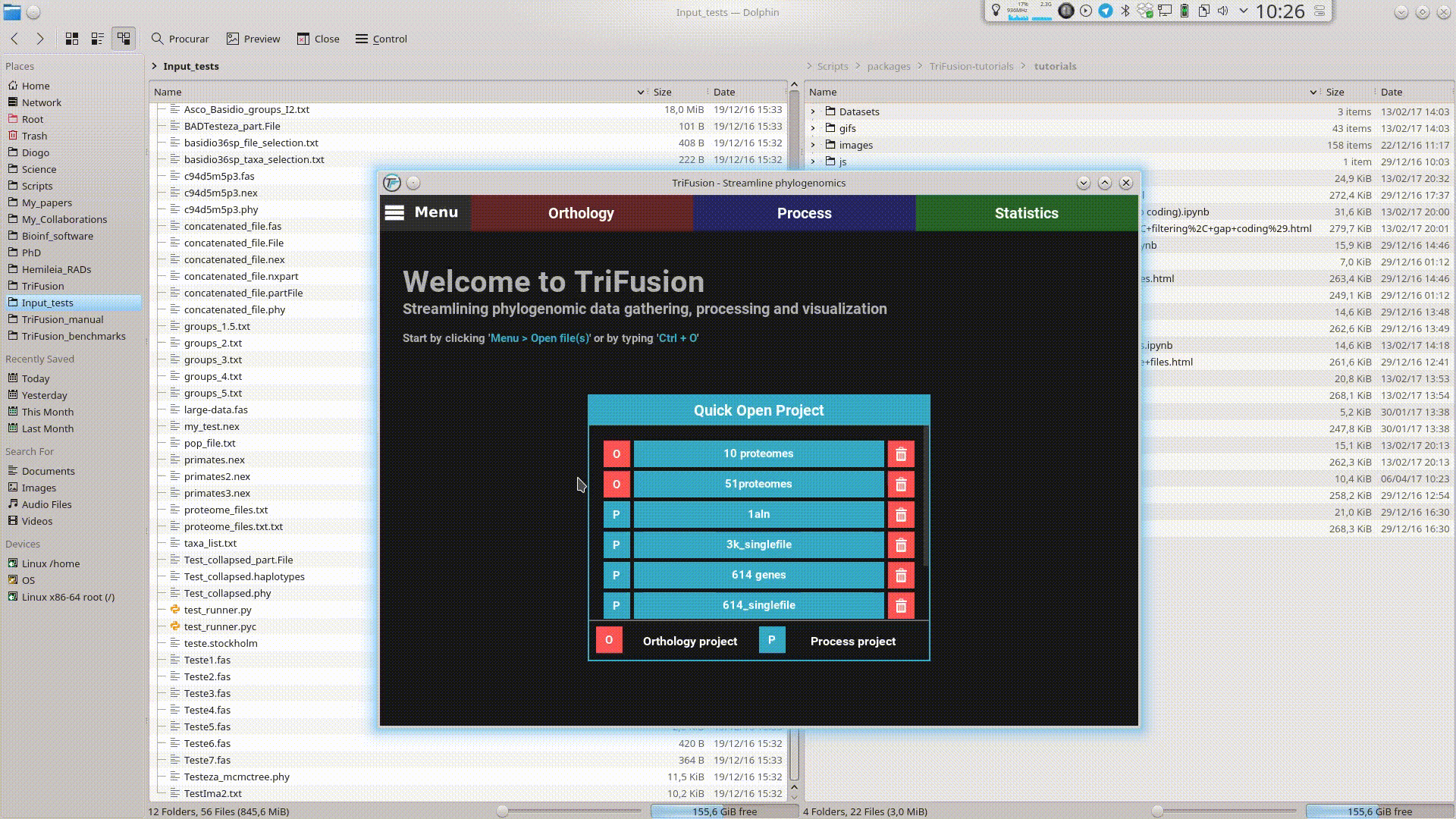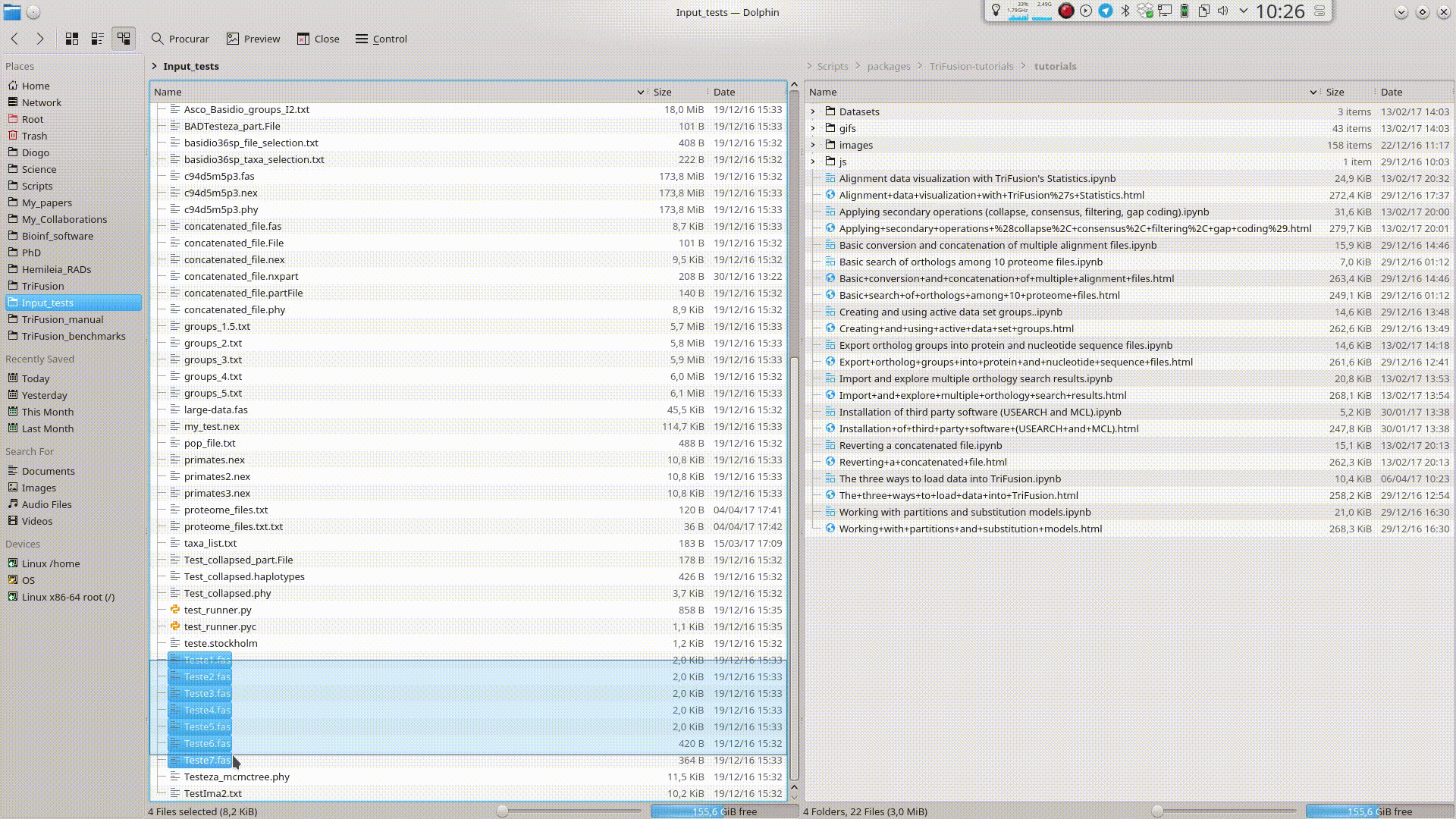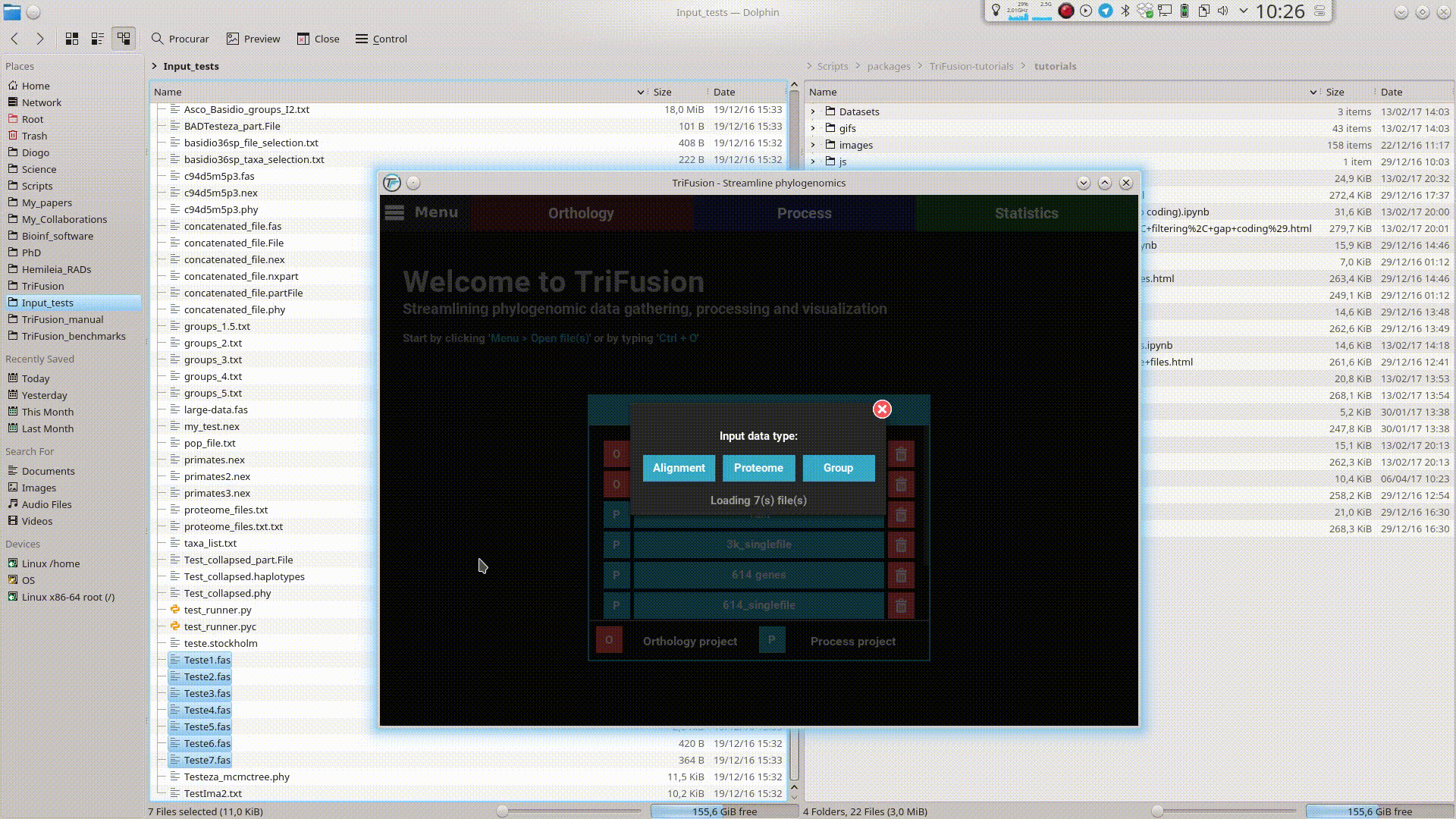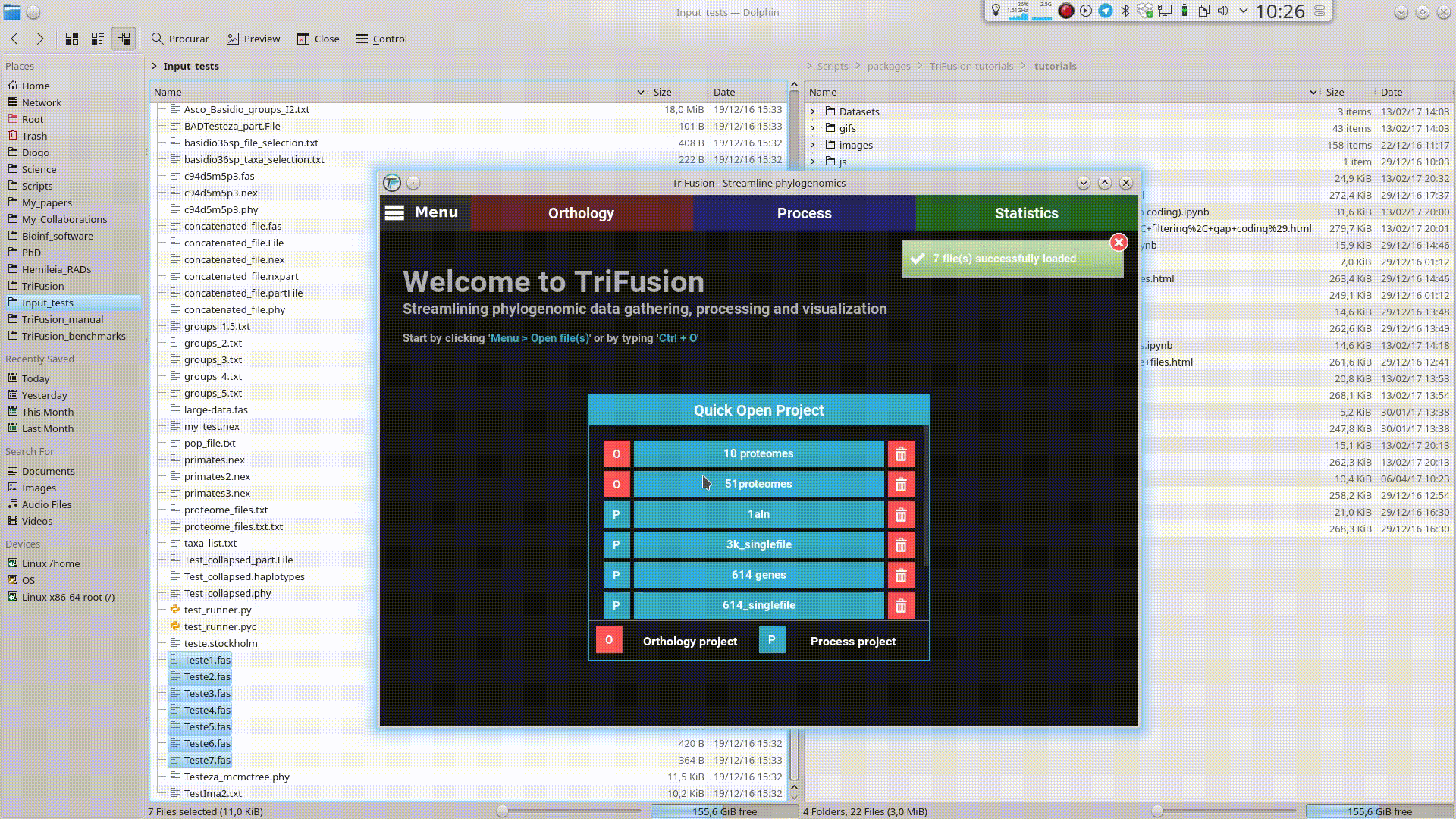
Input files can be provided to TriFusion’s window directly from your systems’ file manager. After selecting the files, drag them into TriFusion’s window, which will display a popup informing of how many files will be loaded and asking whether the files represent alignments, proteomes or groups. Directories can also be dragged as well. In the example below, 7 sequence alignment files are loaded using this method.
Via terminal¶
For terminal lovers (<3) files can be loaded automatically when executing
the TriFusion application. If TriFusion’s executable is already in you
$PATH environmental variable, you can write it in the terminal and then
provide any number of files.
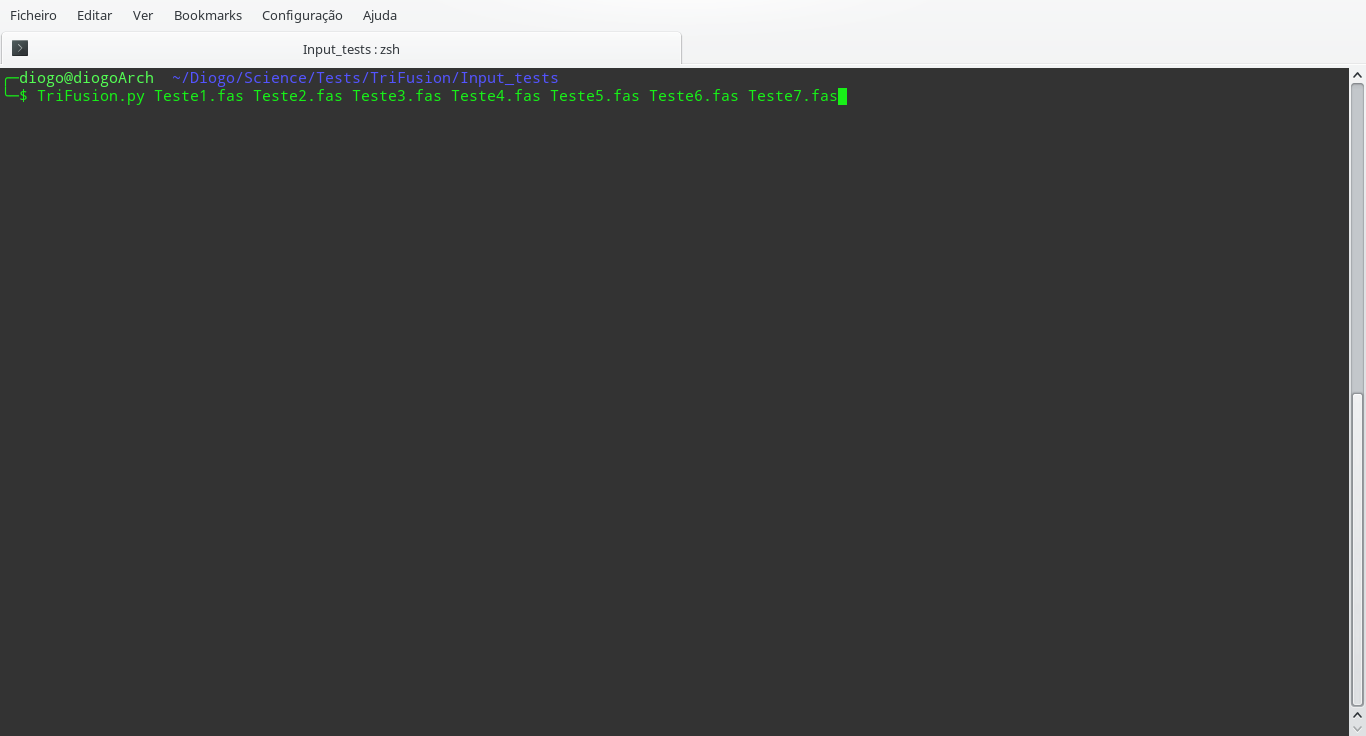
This will open TriFusion and automatically open a popup informing that 7 files will be loaded into TriFusion and asking whether the files represent alignment, proteome or group files. In this case, the data files correspond to alignments.
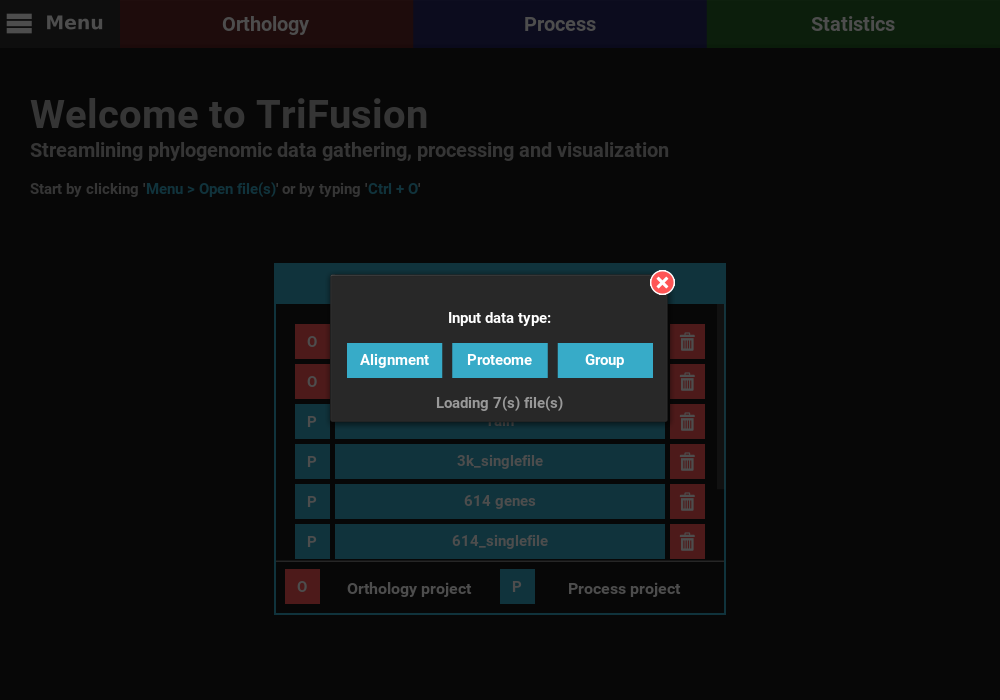
Once the sequence type is selected, the selected files will be loaded normally into TriFusion.